
1
Wyznaczanie stałej Michaelisa-Menten (Km), Vmax oraz określanie typu
inhibicji aktywności fosfatazy kwaśnej
(EC 3.1.3.2 – fosfohydrolaza
monoestrów ortofosforanowych kwaśne opimum).
Cel ćwiczenia
Celem ćwiczenia jest poznanie kinetyki enzymów( v
0
, stałej Km i Vmax) a także określenie
wpływu inhibitorów na szybkość reakcji enzymatycznej.
Wprowadzenie
Fosfataza kwaśna występuje w dużych stężeniach w nasionach roślin. Aktywność
enzymu silnie wzrasta podczas kiełkowania nasion, a następnie maleje w miarę wzrostu
siewek. Prawdopodobnie wzrost jej aktywności związany jest z uwalnianiem fosforanu
nieorganicznego
z
organicznych
form
zapasowych
fosforanu,
np.
z
mezoinozytoloheksafosforanu (kwasu fitynowego).
U zwierząt, fosfataza kwaśna wystepuje głównie w lizosomach komórek należących
do różnych tkanek i narządów. U człowieka, wysokie stężenie enzymu występuje w gruczole
krokowym. Aktywność fosfatazy kwaśnej silnie wzrasta w chorobie nowotworowej tego
gruczołu, co wykorzystuje się w diagnostyce do wczesnego rozpoznawania tej postaci raka.
Aktywność fosfataz oznacza się zazwyczaj używając sztucznych substratów, takich
jak: 2-glicerofosforan, fenylofosforan, fosforan fenoloftaleiny, p-nitrofenylofosforan.
Aktywność enzymu mierzy się ilością uwolnionego fosforanu lub części organicznej estru po
inkubacji enzymu z substratem w ściśle określonych warunkach.
W ćwiczeniu będzie wykorzystany wodny wyciąg enzymu z siewek pszenżyta. Jako
substrat będzie użyty p-nitrofenylofosforan ponieważ jeden z produktów reakcji, p-nitrofenol
po zalkalizowaniu środowiska przechodzi w formę barwną. Ilość wytworzonego p-nitrofenolu
jest miarą aktywności fosfatazy kwaśnej.
1. Badanie kinetyki enzymów
Głównym zadaniem enzymów jest zwiększanie szybkości katalizowanych reakcji, tak
aby mogły sprostać potrzebom metabolizmu. Dla wielu enzymów szybkość reakcji
(v
0
) zmienia się wraz ze zmianą stężenia substratu. Przy wysokim stężeniu substratu
wszystkie cząsteczki enzymu tworzą kompleks enzym-substrat (ES) i wówczas reakcja osiąga
szybkość maksymalną Jest to zjawisko wysycania enzymu substratem. W 1913 roku, na
podstawie badań przebiegu reakcji enzymatycznych, Leonor Michaelis i Maud Menten
sformułowali teorię działania enzymów i ich kinetyki. Zgodnie z tą teorią enzym (E) tworzy
z substratem kompleks enzym-substrat (ES), który następnie rozpada się na: produkt (P)
i wolny enzym (E) według równania:
k
1
k
2
E + S ↔ ES → E + P
k
1
, k
-1
, k
2
– stałe szybkości odpowiednich przemian.
Przy stałym stężeniu enzymu szybkość reakcji zależy od stężenia substratu. Tę zależność
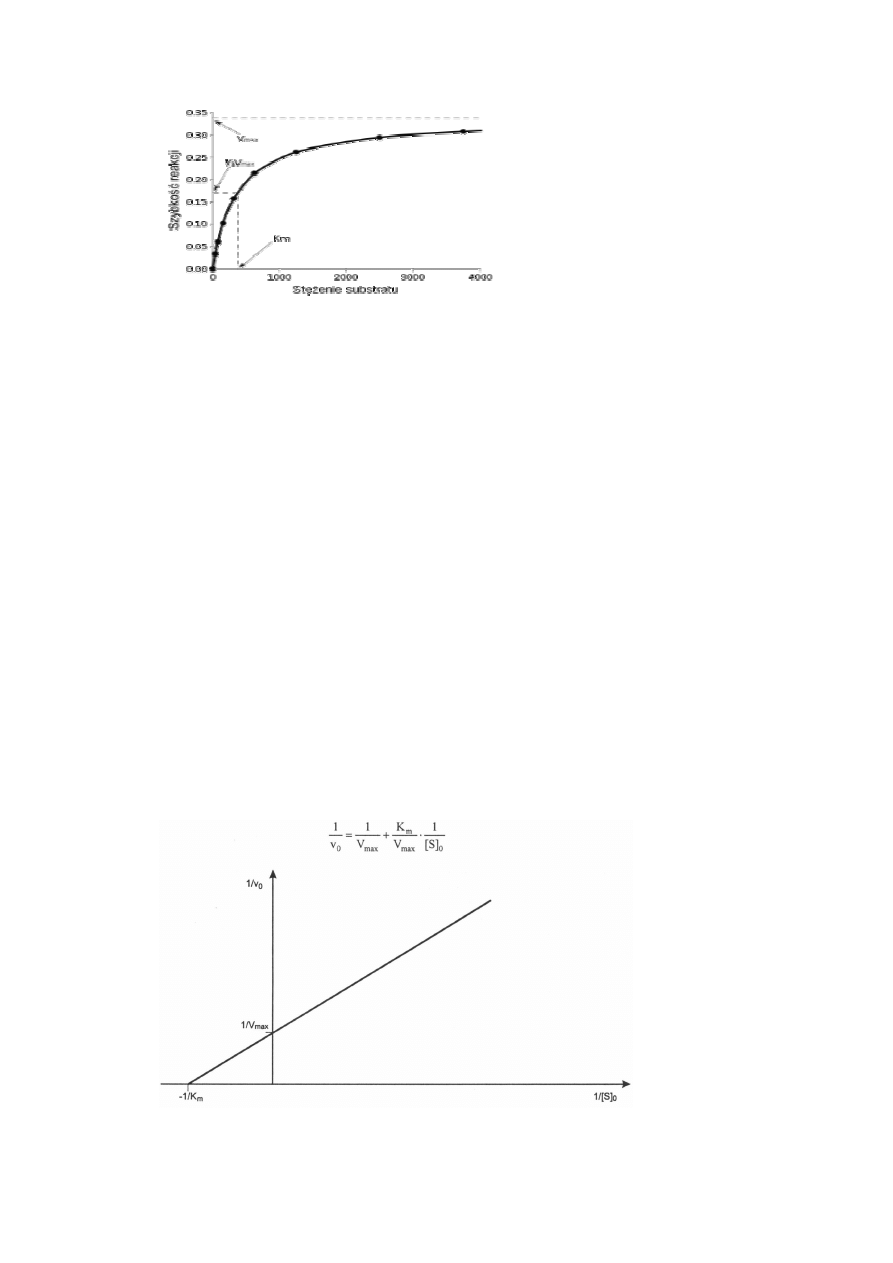
ujmuje równanie Michaelisa-Menten, a obrazuje ją hiperbola (Rys.1).
Vmax [S]
v
0
=
Km + [S]
k
-1
2
Rys.1. Wykres Michaelisa-Menten. Zależność szybkości początkowej reakcji od stężenia
substratu. Km – stała Michaelisa- Menten, Vmax – szybkość maksymalna reakcji.
Przy niewielkim stężeniu substratu, tylko niektóre cząsteczki enzymu tworzą kompleks z
substratem, reakcja przebiega z szybkością v
0
. Przy wysokich stężeniach substratu wszystkie
cząsteczki enzymu połączone są z substratem i reakcja biegnie z szybkością maksymalną
(Vmax). Szybkość reakcji zależy od stężenia kompleksu enzym-substrat [ES] oraz szybkości
rozpadu tego kompleksu, a stałą równowagi tej reakcji jest stała Michaelisa (Km). Km
wyraża się w jednostkach stężenia substratu. Takie stężenie substratu, przy którym szybkość
reakcji stanowi połowę szybkości maksymalnej , nosi nawę stałej Michaelisa (Km).
Dane kinetyczne reakcji takie jak Vmax i Km charakteryzują aktywność metaboliczną
enzymów. Wartość Km oznacza takie stężenie substratu, w którym połowa miejsc aktywnych
w cząsteczkach enzymu jest zajęta, czyli stanowi informację jakie stężenie substratu jest
konieczne do osiągnięcia znaczącego przyspieszenia reakcji. Ponadto, jeśli stała szybkości
rozpadu komplesu ES do substratu i wolnego enzymu jest znacznie wyższa od stałej rozpadu
tego komlpeksu do produktu i enzymu, to wówczas wartość Km jest miarą siły wiązania
substratu przez enzym. Wysoka wartość Km wskazuje na słabe powinowactwo enzymu do
substratu, natomiast niska wartość Km świadczy o silnym wiązaniu substratu przez enzym.
Wyniki doświadczeń wskazują, że dla wielu enzymów Km odpowiada stężeniu substratu in
vivo.
Aby dokładnie wyznaczyć wartość Km oraz Vmax, należy oprzeć się na równaniu
Lineweavera-Burka, które jest odwrotnością równania Michaelisa-Menten, a jego wykresem
jest linia prosta.
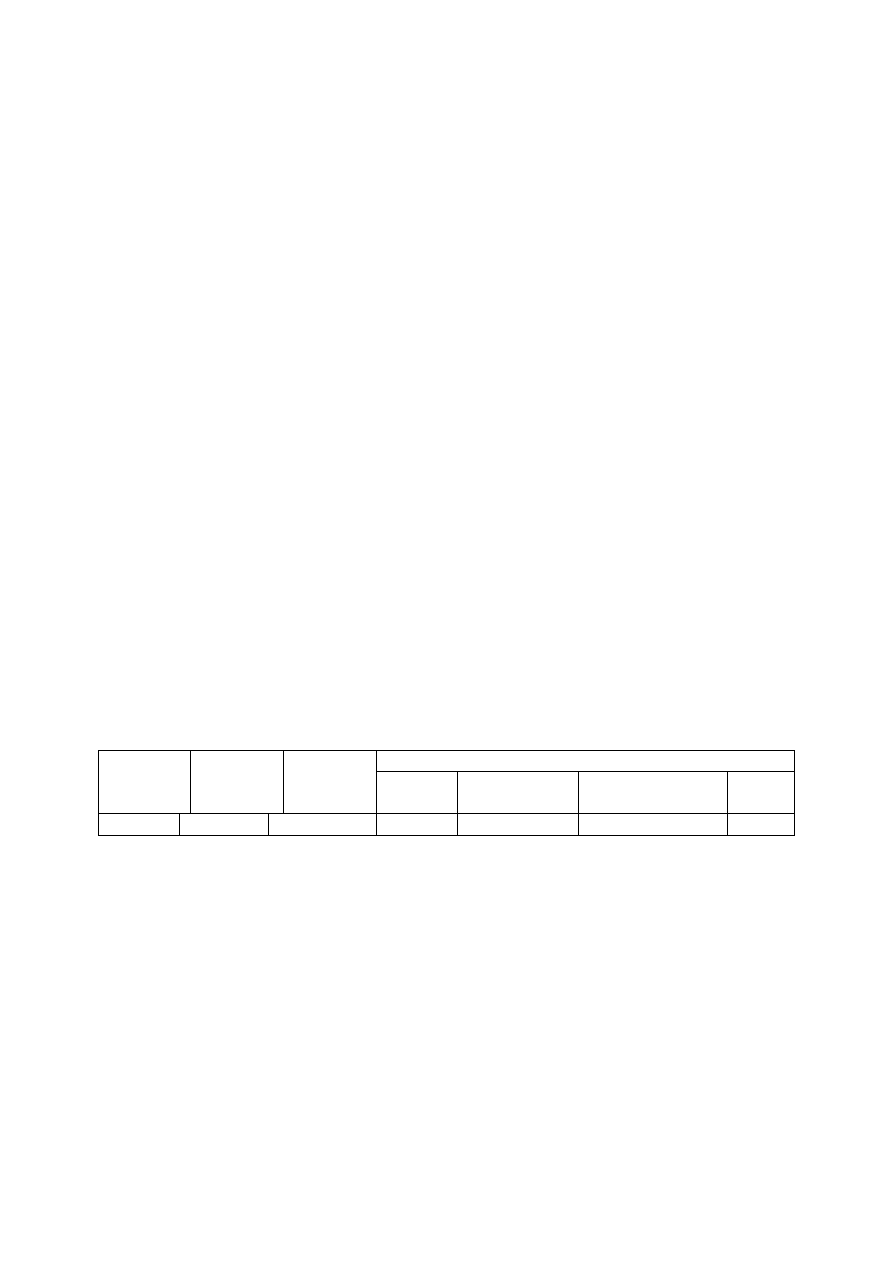
3
Rys.2. Wykres Lineweavera – Burka. Zależność odwrotności szybkości reakcji
enzymatycznej (1/v
0
) od odwrotności stężenia substratu (1/[S]). 1/Vmax – odwrotność
szybkości maksymalnej, 1/Km – odwrotność stałej Michaelisa-Menten.
Odczynniki
1. Wyciąg enzymu: 900 mg liści pszenżyta homogenizować w 100 ml wody
destylowanej przez 5 min (przy wydawaniu prób do kolbek miarowych odpowiednio
zwiększyć ilość liści pszenżyta). Homogenat przesączyć przez sączek z waty i przechowywać
w chłodziarce.
2. 0,0075-molowy 4-nitrofenylofosforan disodowy (substrat).
3. 0,1-molowy bufor octanowy o pH 5,3
4. 10-proc. węglan sodowy.
5. 1,0 mM molibdenian amonu.
Wykonanie (zajęcia 3 godz.)
Wyznaczanie wartości stałej Km dla fosfatazy kwaśnej i oznaczanie aktywności
enzymu.. Do 6 ponumerowanych probówek (próby właściwe) odmierzyć kolejno po 0,05;
0,1; 0,2; 0,3; 0,4 i 0,5 ml p-nitrofenylofosforanu (2), dodać po 0,75 ml buforu octanowego (3)
i uzupełnić wodą do objętości 2,0 ml, a do siódmej probówki (próba kontrolna) odmierzyć
0,75 ml buforu octanowego (3) i 1,25 ml wody destylowanej.
Wszystkie probówki wstawić do łaźni wodnej o temp. 30°C i po ok. 5 min., nie
wyjmując prób z łaźni, dodać kolejno do każdej probówki po 0,5 ml wyciągu enzymu,
wymieszać, zanotować czas, i prowadzić reakcję jeszcze przez 15 min.
Po 15 min przerwać działanie enzymu, dodając kolejno do probówek po 2,5 ml
węglanu sodowego (4), wymieszać i oznaczyć absorbancję w fotometrze przy 420 nm,
ustawionym na zero wobec próby kontrolnej.
Przykładowe obliczenia
Szybkość reakcji = ilość wytworzonego produktu
ml
substratu
[S]
1/[S]
A
420
µ
g produktu =
A
420
/0.002
µ
mole produktu =
µ
g prod./139= v
0
1/v
0
0,1
0,00015
6666
0,132
66
0,475
2,105
Obliczyć ilość µg uwolnionego w reakcji 4-nitrofenolu, dzieląc absorbancję przez
współczynnik absorbancji dla 1µg p-nitrofenolu równy 0,002.. Dla poszczególnych stężeń
substratu obliczyć 1/[S] w mol
–1
· l
–1
, pamiętając, że substrat wyjściowy został rozcieńczony
w mieszaninie inkubacyjnej do 2,5 ml, oraz odwrotność szybkości 1/v (w µmolach produktu).
Sporządzić wykres zależności szybkości reakcji od stężenia substratu, odkładając na osi
rzędnych 1/v (µmole 4-nitrofenolu), a na osi odciętych 1/[S] (mole 4-nitrofenylofosforanu).
Podać prowadzącemu wartość absorbancji dla najwyższego stężenia substratu, a
następnie uwzględniając rozcieńczenia oblicz aktywność fosfatazy kwaśnej wyrażając ją w
µmolach p-nitrofenolu na 1 g liści pszenżyta na 1 min. czasu reakcji.

4
2. Wyznaczanie Km i określanie typu inhibicji fosfatazy kwaśnej przez
molibdenian amonu.
Inhibitory są substancjami zmniejszającymi szybkość reakcji enzymatycznej.
Inhibitorami mogą być metabolity komórkowe, leki, trucizny, jony metali. Zahamowanie
aktywności enzymu w sposób nieodwracalny bądź odwracalny jest jedną z dróg regulacji
szlaków metabolicznych.
Aktywność enzymów nieodwracalnie hamują czynniki fizyczne i chemiczne
powodujące denaturację białka enzymu, albo takie które powodują utlenianie lub alkilowanie
grup funkcyjnych w centrum aktywnym enzymu. Inhibitory nieodwracalne wiążą się
z enzymem za pośrednictwem silnych wiązań kowalencyjnych tworząc nieaktywną
katalitycznie pochodną. Przykładem takiego inhibitora jest diizopropylofluorofosforan (DFP),
związek fosforoorganiczny stosowany jako pestycyd. Tworzy on słabo dysocjujący kompleks
z grupami –OH Ser w miejscu aktywnym esterazy acetylocholiny, enzymu uczestniczącego w
rozkładzie acetylocholiny, mediatora sygnałów nerwowych. Podobnie działa antybiotyk
penicylina, który hamuje aktywność bakteryjnej transpeptydazy peptydoglikanu, enzymu
kluczowego dla syntezy ścian komórkowych.
Inhibicję odwracalną charakteryzuje zdolność do dysocjacji kompleksu enzym-
inhibitor. Do głównych typów inhibicji odwracalnej zaliczamy:
a) inhibicję kompetycyjną;
b) inhibicję niekompetycyjną;
c) inhibicję akompetycyjną ;
d) inhibicję mieszaną.
Inhibitory kompetycyjne (współzawodnicze) są analogami strukturalnymi substratu
i dlatego konkurują z nim o centrum aktywne enzymu. Inhibitor kompetycyjny powoduje
wzrost wartości Km natomiast wartość Vmax pozostaje stała. Przykładem jest hamowanie
reakcji katalizowanej przez dehydrogenazę bursztynianową przy udziale malonianu. Enzym
katalizuje utleniane bursztynianu do fumaranu. Przyłączenie malonianu, do centrum
aktywnego dehydrogenazy bursztynianowej blokuje działanie enzymu przez co nie dochodzi
do uwolnienia produktu. Ten typ inhibicji można cofnąć stosując wysokie stężenia substratu.
Innym przykładem inhibitora kompetycyjnego jest stosowany na ćwiczeniach molibdenian
amonu, który hamuje aktywność fosfatazy kwaśnej.
Inhibitory niekompetycyjne wiążą się odwracalnie z wolnym enzymem (E) lub z
kompleksem enzym-substrat (ES). Substrat oraz inhibitor wiążą się w różnych miejscach
enzymu. Powstający kompleks ESI jest katalitycznie nieaktywny, nie zachodzi uwalnianie
produktu i dlatego wartość Vmax jest niższa w porównaniu do reakcji bez inhibitora. Inhibitor
niekompetycyjny nie zmienia powinowactwa enzymu do substratu i dlatego wartość Km nie
ulega zmianie
Inhibitory akompetycyjne wiążą się odwracalnie z kompleksem enzym-substrat (ES)
tworząc nieaktywny katalitycznie kompleks ESI. W obecności inhibitora akompetycyjnego
obniża się wartość Vmax a także obniża się wartość Km (następuje pozorny wzrost
powinowactwa enzymu do substratu z powodu stabilizacji kompleksu ES). Ten typ inhibicji
najczęściej dotyczy enzymów katalizujących reakcje z udziałem wielu substratów.
Cząsteczki o charakterze inhibitorów mieszanych działają częściowo jak inhibitory
kompetycyjne i niekompetycyjne. Taki inhibitor może przyłączać się zarówno do wolnego E
jak i do kompleksu ES zmniejszając wartość Vmax oraz zwiększając wartość Km. Rodzaj
inhibicji enzymu można określić prowadząc reakcję enzymatyczną przy różnych stężeniach
substratu bez inhibitora oraz w obecności inhibitora a następnie wyznaczając wartości Vmax
oraz Km.
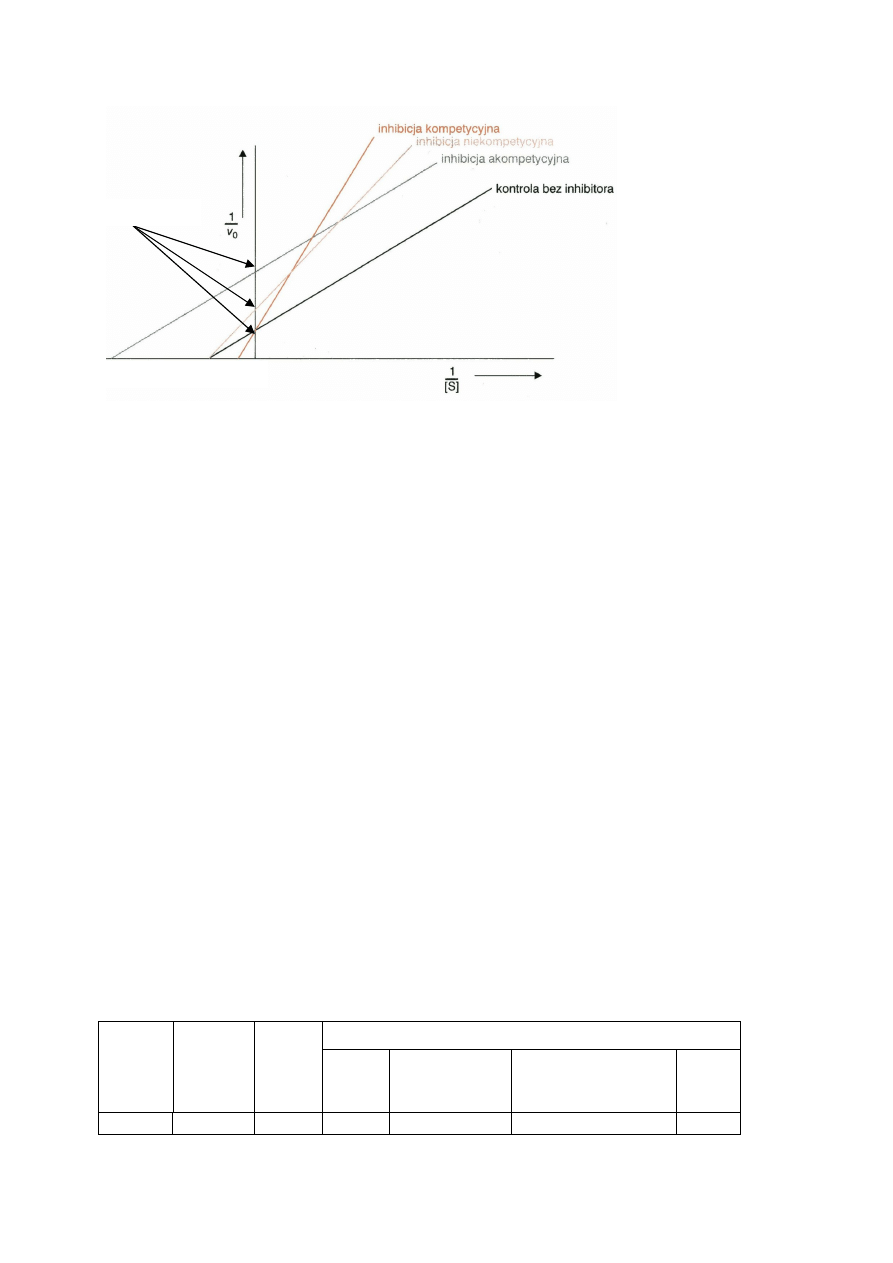
5
Rys. 3. Wykresy Lineweavera – Burka dla reakcji enzymatycznych zachodzących w
obecności inhibitorów.
Różne typy inhibicji można określić przeprowadzając reakcje enzymatyczne przy różnych
stężeniach substratu i inhibitora, a następnie przedstawiając graficznie wyniki analiz.
A. Reakcja bez inhibitora. Do 6 ponumerowanych probówek (próby właściwe)
odmierzyć kolejno 0,05; 0,1; 0,2; 0,3; 0,4 i 0,5 ml p-nitrofenylofosforanu (2), dodać po 0,75
ml buforu octanowego (3) i uzupełnić wodą do objętości 2,0 ml, a do siódmej probówki
(próba kontrolna) odmierzyć 0,75 ml buforu octanowego (3) i 1,25 ml wody destylowanej.
Wszystkie probówki wstawić do łaźni wodnej o temp. 30°C i po ok. 5 min (preinkubacja) nie
wyjmując prób z łaźni, dodać kolejno do każdej probówki po 0,5 ml wyciągu enzymu,
wymieszać, zanotować czas, i prowadzić reakcję jeszcze przez 15 min.
Po 15 min przerwać działanie enzymu, dodając kolejno do probówek po 2,5 ml
węglanu sodowego (4), wymieszać i oznaczyć absorbancję w fotometrze przy 420 nm,
ustawionym na zero wobec próby kontrolnej.
B. Reakcja z inhibitorem. Do 6 ponumerowanych probówek (próby właściwe)
odmierzyć kolejno0,05; 0,1; 0,2; 0,3; 0,4 i 0,5 ml p-nitrofenylofosforanu (2), dodać po 0,75
ml buforu octanowego (3), po 0,05 ml 1mM molibdenianu amonu i uzupełnić wodą do
objętości 2,0 ml, a do siódmej probówki (próba kontrolna) odmierzyć 0,75 ml buforu
octanowego pH 5,3 (3) i 1,25 ml wody destylowanej. Wszystkie probówki wstawić do łaźni
wodnej o temp. 30°C i po ok. 5 min (preinkubacja) nie wyjmując prób z łaźni, dodać
kolejno do każdej probówki po 0,5 ml wyciągu enzymu, wymieszać, zanotować czas, i
prowadzić reakcję jeszcze przez 15 min.
Po 15 min przerwać działanie enzymu, dodając kolejno do probówek po 2,5 ml
węglanu sodowego (4), wymieszać i oznaczyć absorbancję w fotometrze przy 420 nm,
ustawionym na zero wobec próby kontrolnej.
Przykładowe obliczenia
Reakcja bez inhibitora
Szybkość reakcji = ilość wytworzonego produktu
Substrat
(ml)
[S]
1/[S]
A
420
A
420
/0.002 =
µg produktu
Szybkość
reakcji
bez
inhibitora
(v
0
)
=
µ
g
prod./139 =
µmol produktu
1/v
0
0,1
0,00030
3333 0,206
103
0,74
1,35
- 1/Km -1/Km -1/Km
1/Vmax
6
Reakcja w obecności inhibitora
Szybkość reakcji z inhibitorem = ilość produktu w obecności inhibitora
Substrat
(ml)
[S]
1/[S]
A
420
A
420
/0.002 =
µ
g produktu
Szybkość
reakcji
z
inhibitorem
(v
i
)
=
µ
g
prod./139 = µmol produktu
w obecności inhibitora = v
i
1/v
i
0,1
0,00030
3333
0,06
30
0,22
4,5
Opracowanie wyników
1. Obliczyć ilość µg uwolnionego w reakcji 4-nitrofenolu, dzieląc absorbancję przez
współczynnik absorbancji dla 1µg p-nitrofenolu równy 0,002. Dla poszczególnych stężeń
substratu obliczyć 1/[S] w mol
–1
· l
–1
, pamiętając, że substrat wyjściowy został rozcieńczony
w mieszaninie inkubacyjnej do 2,5 ml, oraz odwrotność szybkości 1/v
0
dla reakcji bez
inhibitora oraz 1/v
i
dla reakcji z inhibitorem(w µmolach produktu). Sporządzić wykres
zależności szybkości reakcji od stężenia substratu, odkładając na osi rzędnych 1/v
0
lub 1/v
i
(µmole p-nitrofenolu), a na osi odciętych 1/[S] (mole p-nitrofenylofosforanu). Przedłużyć
linię do przecięcia się z ujemną częścią osi x, z otrzymanej wartości –1/K
m
obliczyć stałą K
m
.
2. Podać prowadzącemu wartość absorbancji dla najwyższego stężenia substratu, a
następnie uwzględniając rozcieńczenia oblicz aktywność fosfatazy kwaśnej wyrażając ją w
µmolach p-nitrofenolu na 1 g liści pszenżyta na 1 min. czasu reakcji.
3. Obliczyć ilość µg uwolnionego w reakcji p-nitrofenolu, dzieląc absorbancję przez
współczynnik absorbancji dla 1µg p-nitrofenolu równy 0,002. Sporządzić wykres zależności
szybkości
reakcji
od
stężenia.
Dla
poszczególnych
stężeń
substratu
obliczyć
1/[S] w mol
–1
· l
–1
, pamiętając, że substrat wyjściowy został rozcieńczony w mieszaninie
inkubacyjnej do 2,5 ml, oraz odwrotność szybkości 1/v (w µmolach produktu). Sporządzić
wykres zależności szybkości reakcji bez inhibitora i z inhibitorem od stężenia substratu,
odkładając na osi rzędnych odpowiednio 1/v
0
i 1/v
i
(µmole p-nitrofenolu), a na osi odciętych
1/[S] (mole p-nitrofenylofosforanu).
Przedłużyć linię do przecięcia się z ujemną częścią osi x, z otrzymanej wartości –1/K
m
obliczyć wartość stałej Km dla reakcji niehamowanej i reakcji hamowanej.
4. Obliczyć wartość Vmax wiedząc, że punkt przecięcia prostej z osią OY wyznacza
wartość 1/Vmax.
Szybkość maksymalną dla reakcji bez inhibitora można obliczyć ze wzoru: V
max
= v
0
(1+ K
m
/[S]) mając wyznaczoną wartość K
m
i aktualne stężenie substratu oraz dla reakcji
w obecności inhibitora ze wzoru: V
max,i
= v
i
(1+ K
m,i
/[S]) mając wyznaczoną wartość K
mi
i aktualne stężenie substratu.
5. Ustalić typ inhibicji analizując otrzymane wykresy a także uzyskane wartości
Vmax,i oraz Km,i.
Pytania
1.
Co to jest stała Michaelisa-Menten i od czego zależy jej wartość.
2.
Podać równanie Lineweavera-Burka i omówić wyznaczanie stałej K
m
dla fosfatazy
kwaśnej w oparciu o postępowanie Lineweavera-Burka.
3.
Omówić krótko działanie inhibitora kompetycyjnego oraz metodę określania typu
inhibicji aktywności enzymu.
4.
Oblicz wartość V
max
wiedząc, że:
Km=1,19 x 10
-3
M;
v
0
= 0,85
[S] = 7,5 x 10
-3
M

7
5.
Po przeprowadzeniu reakcji enzymatycznej bez inhibitora oraz z inhibitorem
uzyskano następujące wyniki: Km= 2,6 x 10
-4
M, Vmax = 1,25 x 10
-5
M, a dla
reakcji z inhibitorem Kmi = 4,2 x 10
-3
M oraz Vmax = 1,25 x 10
-5
M. Na jaki typ
inhibicji wskazują dane doświadczalne.
6.
Sporządzono 500 ml 0,4 % roztworu p-nitorofenylofosforanu disodowego. Oblicz
stężenie molowe tego roztworu.
Literatura
1.
Berg, J.M., Tymoczko J.L., Stryer L. Biochemia. Wydawnictwo Naukowe PWN 2009
2.
Witwicki J. Ardelt W. Elementy enzymologii PWN 1984
3.
Senna R., Simonin V., Silva-Neto M.A.C., Fialho E.. 2006. Induction of acid
phosphatase activity during germination of maize (Zea mays) seeds. Plant Phys.
Bioch. 44, 467-473.
4.
Cashikar A. G., Kumaresan R., Rao M. 1997 Biochemical characterization and
subcellular localization of the kidney bean purple acid phosphatase. Plant Physiol. 11;
907-913.
Wyszukiwarka
Podobne podstrony:
Biochemia(ŻCz)Ćw3 Wyznaczanie stałej Michaelisa Km
HYDROLIZA SACHAROZY – WYZNACZANIE STAŁEJ MICHAELISA
WYZNACZANIE STAŁEJ PLANCKA ORAZ PRACY WYJŚCIA ELEKTRONU
Wyznaczanie Km i Vmax
Pomiary pH roztworów oraz wyznaczanie stałej dysocjacji słabego kwasu Ćw 4
5 Wyznaczanie stałej Plancka oraz pracy wyjścia elektronu
20. Wyznaczanie stałej Planck oraz pracy wyjścia elektronu
WYZNACZANIE STAŁEJ PLANCKA ORAZ PRACY WYJŚCIA ELEKTRONU
Wyznaczanie Km i Vmax
Pomiary pH roztworów oraz wyznaczanie stałej dysocjacji słabego kwasu Ćw 4
Sprawozdanie 2 Wyznaczanie stałej Planca oraz pracy wyjścia elektronu (2)
5 Wyznaczanie stałej Plancka oraz pracy wyjścia elektronu docx
Ćw 03c Izolacja limfocytów ze śledziony oraz określanie żywotności komórek
Wyznaczanie stałej siatki dyfrakcyjnej, Prz inf 2013, I Semestr Informatyka, Fizyka, SPRAWOZDANIA DU
Wyznaczanie stałej reakcji szybkości zmydlania estru, Studia, Politechnika
302 Wyznaczanie stałej siatki dyfrakcyjnej
więcej podobnych podstron